2026年5月6日 发表于新英格兰医学杂志
摘要
背景
目前针对胰腺导管腺癌 (PDAC) 患者的治疗方法疗效有限。超过 90% 的 PDAC 肿瘤存在RAS激活突变。Daraxonrasib (RMC-6236) 是一种口服 RAS(ON) 多选择性抑制剂,可靶向鸟苷三磷酸结合的突变型和野生型 RAS。
方法
在这项 I/II 期研究中,我们评估了 daraxonrasib 在携带RAS激活突变的晚期实体瘤患者中的疗效。患者每日口服一次 10 至 400 mg daraxonrasib;300 mg 被选为 III 期剂量。主要终点是安全性。药代动力学和抗肿瘤活性是次要终点。本报告重点关注 168 例既往接受过治疗的RAS突变型胰腺导管腺癌 (PDAC) 研究患者。
结果
在接受 daraxonrasib(剂量≤300 mg)治疗的168例胰腺导管腺癌(PDAC)患者中,96%报告了任何级别的治疗相关不良事件;其中30%报告了3级或以上不良事件。发生率≥10%的治疗相关不良事件包括皮疹、腹泻、恶心、口腔炎或黏膜炎、呕吐和疲乏。在接受二线 daraxonrasib(剂量≤300 mg)治疗的26例RAS G12突变患者亚组中,35%的患者报告了客观缓解(95%置信区间[CI]:17%~56%)。中位缓解持续时间为8.2个月(95% CI:3.8个月至无法评估),中位无进展生存期为8.5个月,中位总生存期为13.1个月。在38例RAS G12、G13或Q61突变患者中,29%(95% CI,15%至46%)达到客观缓解。中位缓解持续时间为8.2个月(95% CI,3.8至8.8个月),中位无进展生存期为8.1个月,中位总生存期为15.6个月。
结论
在既往接受过治疗的RAS突变型胰腺导管腺癌(PDAC)患者中,daraxonrasib 与三分之一的患者出现3级或以上治疗相关不良事件相关;同时,也报道了其抗肿瘤活性。(由Revolution Medicines公司资助;RMC-6236-001;ClinicalTrials.gov注册号:NCT05379985)
正文
胰腺导管腺癌(PDAC)是一种高度致命的癌症,大多数患者在确诊时已处于晚期。对于转移性疾病患者,中位总生存期不足一年,5年后仅有3%的患者存活。除少数适合靶向治疗的患者外,转移性PDAC的标准治疗方案主要依赖于以5-氟尿嘧啶或吉西他滨为基础的化疗。二线化疗方案疗效有限,客观缓解率(即确认的完全缓解或部分缓解)低于10%,中位总生存期仅为5至7个月。
编码大鼠肉瘤病毒( RAS )的基因家族经典成员——即KRAS、HRAS和NRAS——的致癌突变存在于超过90%的胰腺导管腺癌(PDAC)肿瘤中,其中大多数突变(>80%)是KRAS第12密码子的氨基酸替换。在小鼠模型中,KRAS突变启动PDAC的发生,而突变蛋白的清除则可诱导肿瘤消退。甘氨酸12(G12)、甘氨酸13(G13)和谷氨酰胺61(Q61)的突变会损害鸟苷三磷酸酶(GTP酶)的活性,从而导致活性RAS蛋白与GTP结合并持续激活致癌信号通路。
长期以来,人们一直认为使用药物靶向RAS是无法实现的,但共价KRAS G12C(OFF)抑制剂的开发——这些抑制剂通过共价结合将KRAS G12C突变蛋白锁定在“关闭”状态——证明了直接靶向RAS是可行的。然而,KRAS G12C突变在胰腺导管腺癌(PDAC)中较为罕见(发生率仅为1%至2%),这限制了这些药物在该疾病中的临床应用。此外,由于PDAC中的大多数RAS突变通过GTP结合(ON)状态驱动组成型信号传导,因此RAS(OFF)抑制剂对激活的RAS的抑制通常不完全。这些抑制剂的疗效还受到耐药性的进一步限制,包括继发性RAS突变的出现。因此,需要开发靶向活性GTP结合RAS并针对不同RAS变体的疗法。
Daraxonrasib (RMC-6236) 是一种口服生物利用度高的 RAS(ON) 多选择性非共价抑制剂,对突变型和野生型 KRAS、HRAS 和 NRAS 的活性状态均有抑制作用。Daraxonrasib 通过在细胞内与环孢菌素 A 和 GTP 结合的 RAS 形成三元复合物,从空间位阻上阻断 RAS 效应蛋白的结合,从而强效抑制下游信号传导。临床前研究表明,daraxonrasib 对RAS突变型癌症具有深度且持久的疗效,尤其在 12 号密码子突变的肿瘤中疗效最为显著。这些研究结果包括在胰腺癌模型中观察到明显的肿瘤消退。在一项先前对这项多中心、开放标签的 I/II 期研究的分析中,纳入了携带RAS突变的晚期实体瘤患者,111 例患者中有 16 例 (14.4%) 发生了 3 级或以上的治疗相关不良事件,并观察到了初步的抗肿瘤活性。在此,我们报告了 RMC-6236-001 研究的结果,该研究评估了 daraxonrasib 在既往接受过治疗的RAS突变型晚期 PDAC 患者中的安全性、剂量优化水平和抗肿瘤活性。
方法
患者
符合条件的患者为年龄≥18岁的成年患者,且患有携带KRAS、NRAS或HRAS基因12、13或61号密码子突变的晚期实体瘤。胰腺导管腺癌(PDAC)患者若符合以下条件则符合入组条件:接受氟尿嘧啶或吉西他滨类化疗后出现疾病进展或不可耐受的副作用;根据实体瘤疗效评价标准(RECIST)1.1版评估,存在可测量病灶;且美国东部肿瘤协作组(ECOG)体能状态评分为0或1分(采用5分制,分数越高表示残疾程度越重)。主要排除标准包括未经治疗的中枢神经系统转移和既往接受过RAS靶向治疗。完整的入组标准详见研究方案。
治疗
在剂量递增阶段,RAS突变型实体瘤患者接受daraxonrasib 每日一次治疗,21天为一个周期,剂量范围为10 mg至400 mg。采用贝叶斯分析法确定最大耐受剂量(MTD)和II期推荐剂量。尽管400 mg剂量下频繁的剂量调整限制了 daraxonrasib 持续给药的可行性,但并未正式达到MTD。
KRAS G12突变型胰腺导管腺癌(PDAC)的扩展队列分别以120 mg、200 mg或300 mg的剂量入组,而携带RAS G12以外其他突变的PDAC患者则以300 mg的剂量入组于另一个队列。由于终点或入组标准不同,本报告排除了三个探索性300 mg队列(包括接受强制性活检、RAS野生型肿瘤或既往未接受过RAS突变型PDAC治疗的患者)中的患者数据。治疗持续至出现不可接受的毒性反应、疾病进展、撤回知情同意或研究终止(以先发生者为准)。符合预设标准的患者可在疾病进展后继续接受治疗。
终点和评估
主要目标是评估安全性和副作用。本报告总结了研究者根据美国国家癌症研究所不良事件通用术语标准(NCI-CTCAE)第5版对 daraxonrasib 治疗相关的不良事件进行分级。关键次要终点包括初步抗肿瘤活性和药代动力学。抗肿瘤活性终点包括客观缓解率、缓解持续时间和无进展生存期,由研究者根据RECIST 1.1版进行评估。根据方案规定,在基线时以及治疗结束后每2至3个周期进行影像学检查,直至治疗结束。探索性终点包括总生存期以及对血浆(Guardant Infinity检测)和肿瘤组织(Tempus xE检测)中同时存在的基因组改变的分析。
安全性和有效性
所有既往接受过转移性胰腺癌治疗且至少接受过一次daraxonrasib(剂量≤300 mg)治疗的患者均纳入安全性和有效性评估,无论该药物是在剂量递增阶段还是剂量优化阶段给药。接受剂量≤120 mg的患者被归为一组进行分析。由于暴露时间存在重叠,接受剂量160至220 mg的患者被归为一组;300 mg是剂量优化组中评估的最高剂量。
由于临床前模型表明RAS G12 突变肿瘤对daraxonrasib 最为敏感,因此,研究人员根据该突变的存在情况,对亚组患者进行了预先设定的疗效描述性分析。这些患者也被纳入了RAS突变肿瘤(G12、G13 和 Q61 位点均有氨基酸替换)的分组分析。此外,研究人员还根据治疗线数进行了疗效分析,其中二线治疗定义为既往接受过一线转移性疾病治疗,三线及以上治疗定义为既往接受过两线或以上转移性疾病治疗。
统计分析
我们使用描述性统计方法总结了患者的基线特征、安全性和疗效。未进行正式的统计假设检验。在适当情况下,计算了双侧 95% 置信区间。我们使用 Clopper-Pearson 精确法估计了比例的置信区间,并使用 Kaplan-Meier 法估计了中位无进展生存期、缓解持续时间和总生存期。双侧 95% 置信区间采用 Brookmeyer 和 Crowley 法计算。
结果
患者
从 2022 年 6 月 22 日至 2025 年 6 月 30 日,共有 168 例RAS突变型 PDAC 患者接受了至少一剂daraxonrasib 作为二线(或后续)治疗:83 例患者接受了 300 mg 的剂量,51 例患者接受了 160 至 220 mg 的剂量,34 例患者接受了 120 mg 或更少的剂量(见补充附录图S1)。
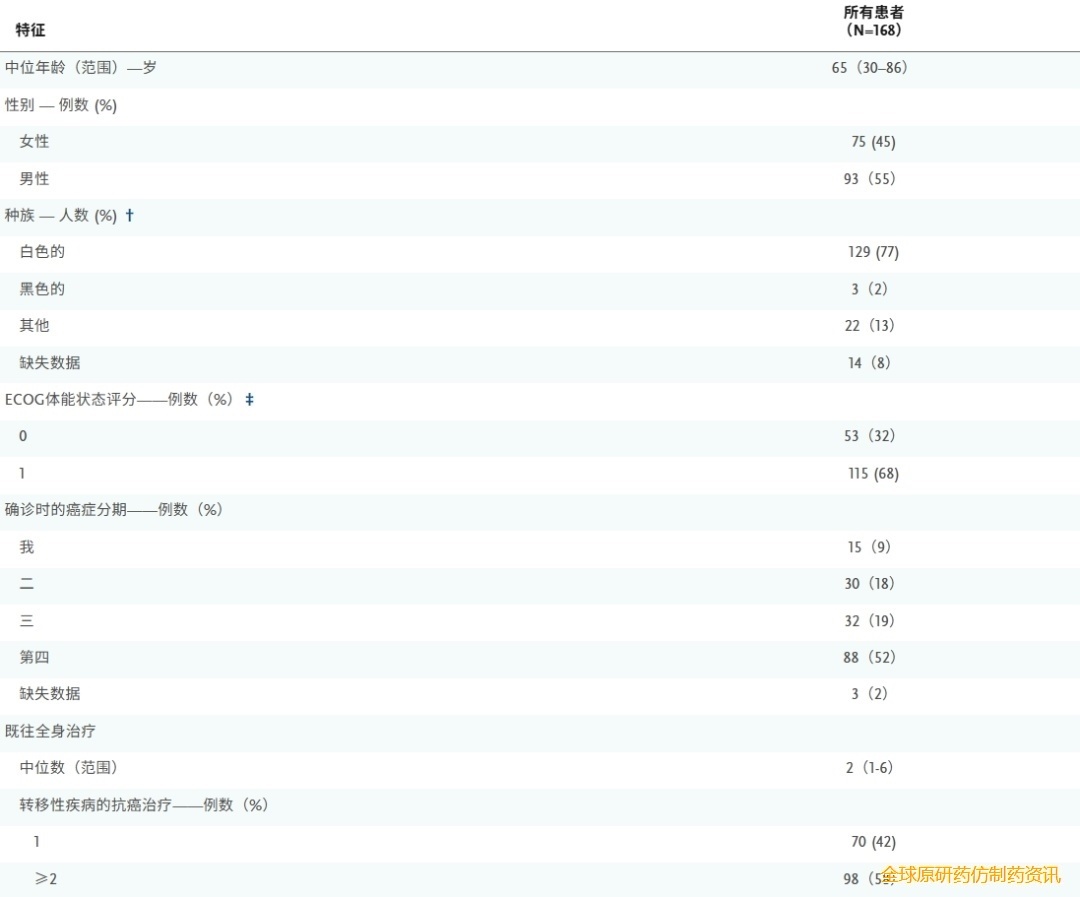
表1总结了患者基线时的人口统计学和临床特征。所有剂量组的中位年龄为65岁(范围30至86岁),45%的患者为女性。68%的患者ECOG体能状态评分为1分。所有患者入组时均为IV期,其中67%的患者出现肝转移,46%的患者出现肺转移。
表1 患者基线特征
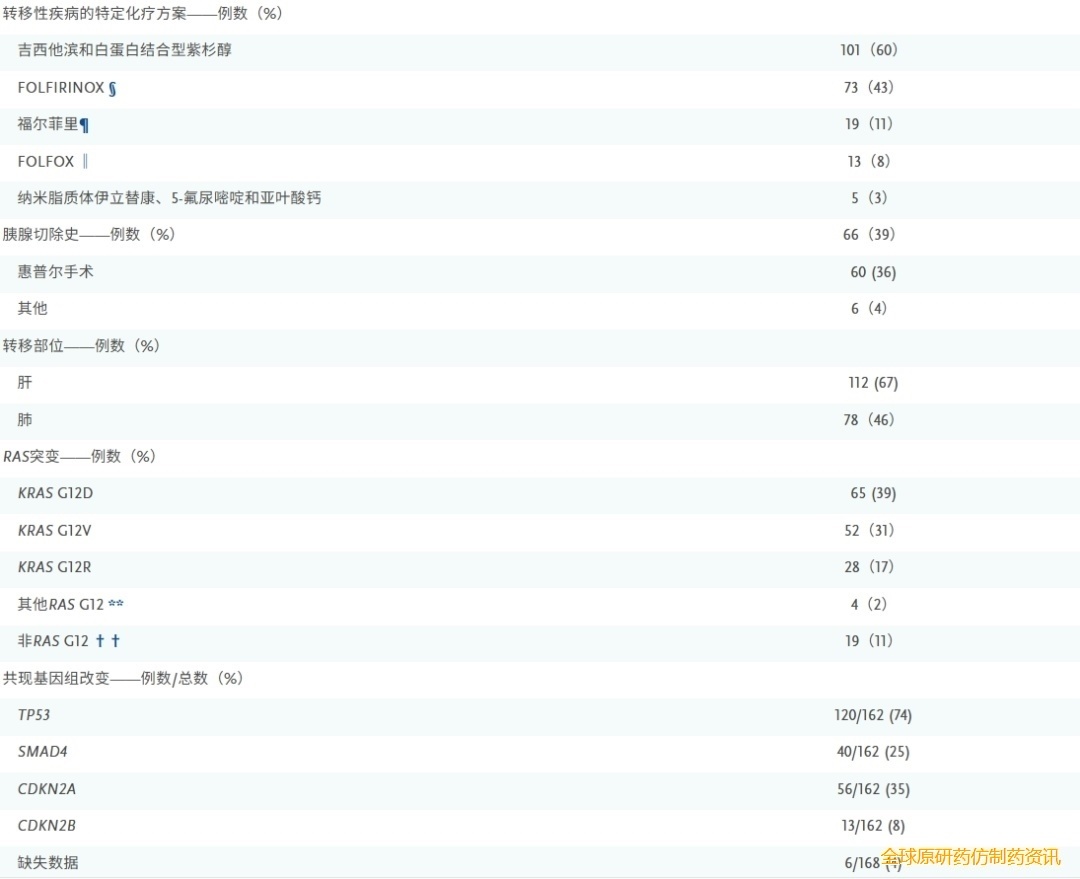
在168例患者中,149例(89%)存在RAS G12突变。这些突变包括KRAS G12D(39%)、KRAS G12V(31%)、KRAS G12R(17%)和其他RAS G12突变(2%)。在19例非RAS G12突变患者中,14例存在KRAS Q61H突变,2例存在KRAS Q61R突变,2例存在KRAS Q61K突变,1例存在KRAS G13D突变。TP53 (74%)、SMAD4(25%)、CDKN2A(35%)和CDKN2B (8%)是常见的共存基因组改变。这些突变的发生率与PDAC的预期患病率一致。
所有患者既往接受全身治疗的中位数为2种(范围:1至6种)。70例患者(42%)接受过1线治疗,98例患者(58%)接受过2线或2线以上转移性疾病治疗。既往转移性疾病治疗方案包括吉西他滨联合白蛋白紫杉醇(60%的患者)和FOLFIRINOX方案(由亚叶酸钙、氟尿嘧啶、伊立替康和奥沙利铂组成)或改良FOLFIRINOX方案(43%的患者)。接受300 mgdaraxonrasib 作为二线、三线或更远线治疗的患者的基线特征总结于表S1。
在安全性分析人群的168例患者中,治疗持续时间中位数为5.7个月(范围:0.03至31.5个月);接受300 mg剂量治疗的患者中,治疗持续时间中位数为5.8个月(范围:0.03至22.3个月);接受160至220 mg剂量治疗的患者中,治疗持续时间中位数为6.0个月(范围:0.4至25.5个月);接受120 mg或更低剂量治疗的患者中,治疗持续时间中位数为5.1个月(范围:0.2至31.5个月)。168例患者中有150例停止治疗,最常见的原因是疾病进展(93例[55%])、临床进展(21例[12%])或不良事件(8例[5%])。
安全
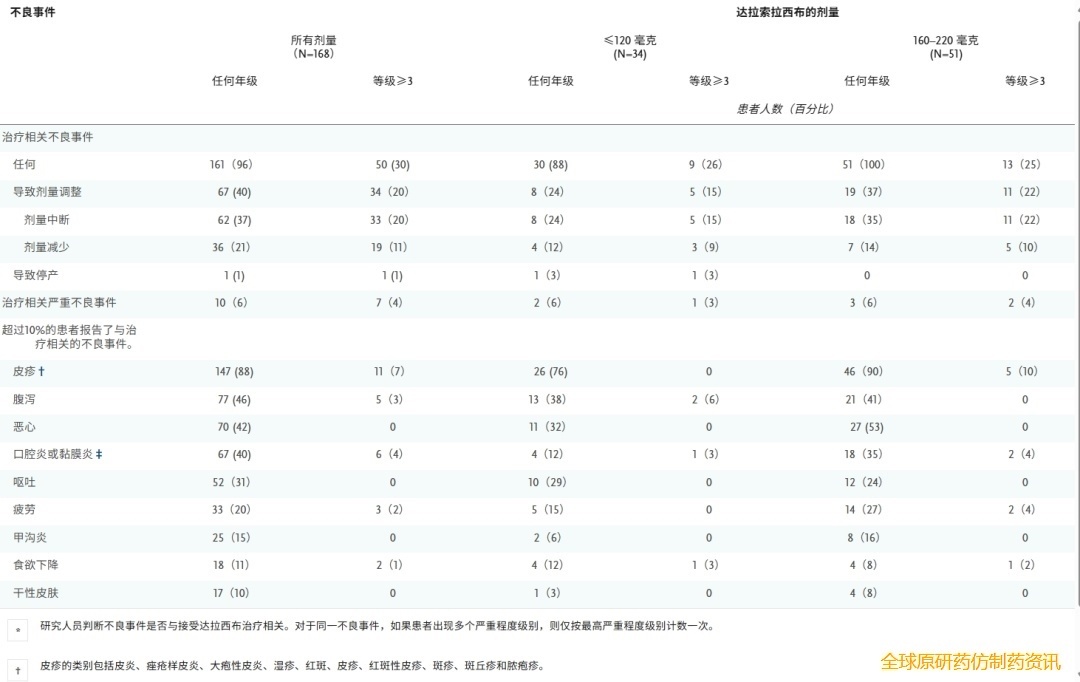
在纳入的168例患者的安全性分析人群中,166例患者(99%)在治疗期间观察到任何级别的不良事件,无论其归因如何。最常见的不良事件(发生率≥30%)为皮疹(89%)、腹泻(50%)和恶心(47%)。110例患者(65%)报告了3级或更高级别的不良事件。
研究者认为与 daraxonrasib 治疗相关的不良事件在96%的患者中报告;其中大多数为1级或2级(表2)。最常见的治疗相关不良事件(任何级别)为皮疹(88%)、腹泻(46%)和恶心(42%)。30%的患者发生3级或以上治疗相关不良事件,未观察到5级不良事件。6%的患者发生严重治疗相关不良事件,其中腹泻(2%)最为常见。未达到最大耐受剂量(MTD),最大给药剂量为400 mg。治疗相关不良事件的剂量相关性增加以及剂量调整支持选择300 mg剂量进行进一步评估。
在接受 300 mg 剂量治疗的 83 例患者中,96% 的患者出现了任何级别的治疗相关不良事件,最常见的不良事件为皮疹(90%)、口腔炎或黏膜炎(54%)、腹泻(52%)和恶心(39%)。34% 的患者出现了 3 级或更高级别的不良事件,其中皮疹和贫血的发生率均为 7%。这些不良事件导致 48% 的 300 mg 剂量组患者调整了剂量,包括 43% 的患者暂停用药,30% 的患者减少剂量。导致超过 10% 的患者调整剂量的任何级别不良事件均为皮疹和口腔炎或黏膜炎(表S2)。皮疹和胃肠道不良反应可通过常规临床干预措施进行控制,例如局部应用糖皮质激素和抗生素(如克林霉素)、全身应用抗生素(多西环素或米诺环素)、防晒措施、止泻治疗以及剂量调整(表S3)。在300 mg组中,13%的患者出现3级或以上治疗相关不良事件,导致剂量减少;部分剂量减少是由于1级或2级不良事件(如皮疹、口腔炎或黏膜炎)引起的。由治疗相关不良事件导致的累积剂量中断的中位持续时间为16天。300 mg组的平均相对剂量强度为86%,且无患者因这些不良事件而停止治疗。当需要中断剂量时,大多数患者能够以相同或更低的剂量恢复治疗,从而维持总体剂量强度。
表2. RAS突变型 PDAC患者的安全性总结。
药代动力学
daraxonrasib 具有口服生物利用度,且其暴露量呈剂量依赖性增加(图S2)。daraxonrasib 稳态全血浓度曲线下面积在80 mg至300 mg剂量范围内近似呈剂量比例关系,重复给药后药物蓄积极少。根据初步群体药代动力学模型,daraxonrasib 达到最大血药浓度的中位时间为1.6至3.9小时,平均末端半衰期为11.8小时(变异系数为28%)。由于160 mg和200 mg至220 mg剂量组的暴露量存在显著重叠,因此将这两个剂量组的数据合并,以便进行有效的安全性和有效性评估。
功效
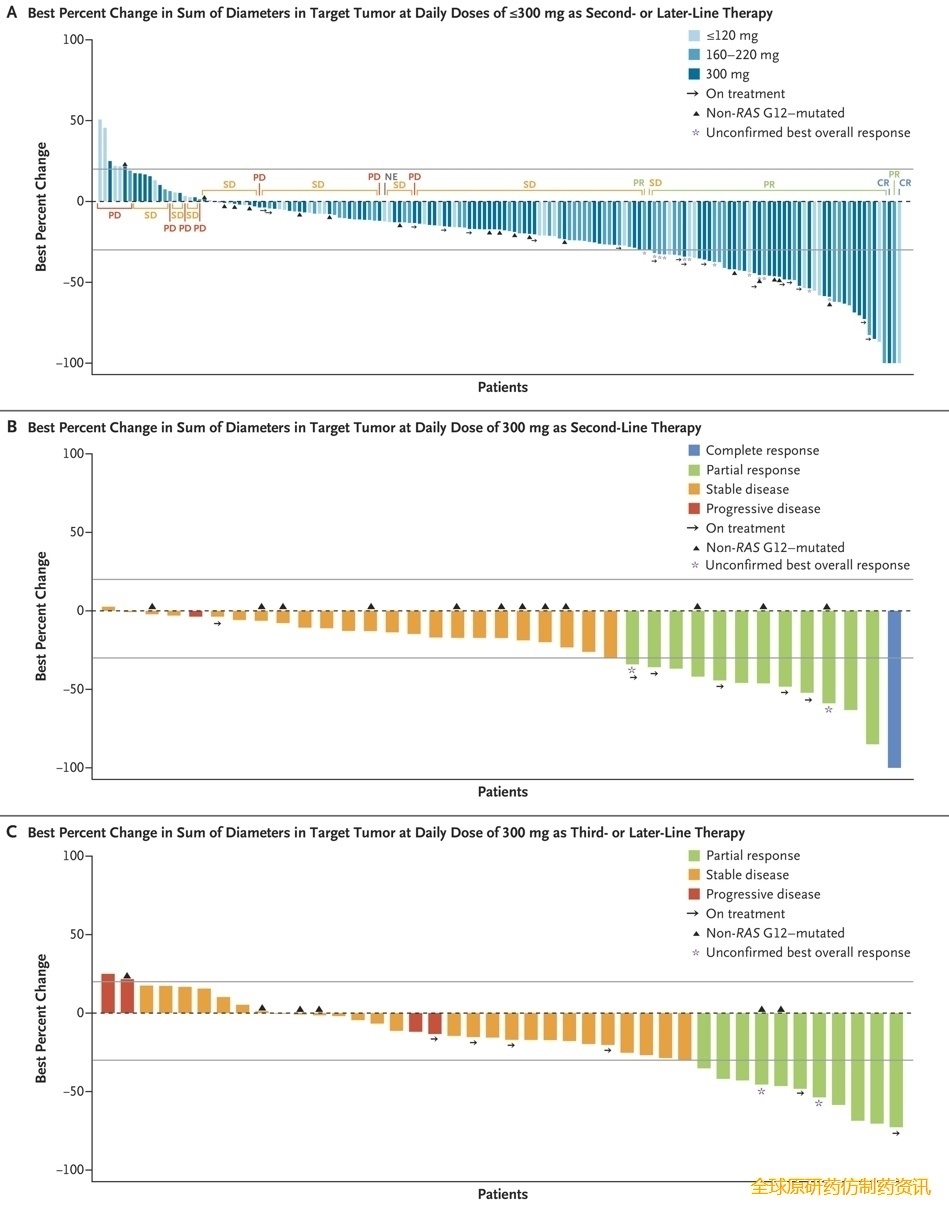
Daraxonrasib 在 300 mg 或更低剂量下以及针对多种RAS突变均显示出抗肿瘤活性(图1A)。300 mg 剂量组的缓解率通常高于较低剂量组,且在早期治疗中疗效更佳(表S4)。作为二线治疗,300 mg 剂量组在 6 个月和 9 个月时的无进展生存期和总生存期里程碑事件发生率均高于较低剂量组(表S5和S6)。这些疗效数据也支持对 300 mg 剂量进行进一步评估。
图1. Daraxonrasib 在RAS突变型 PDAC 中的抗肿瘤活性。
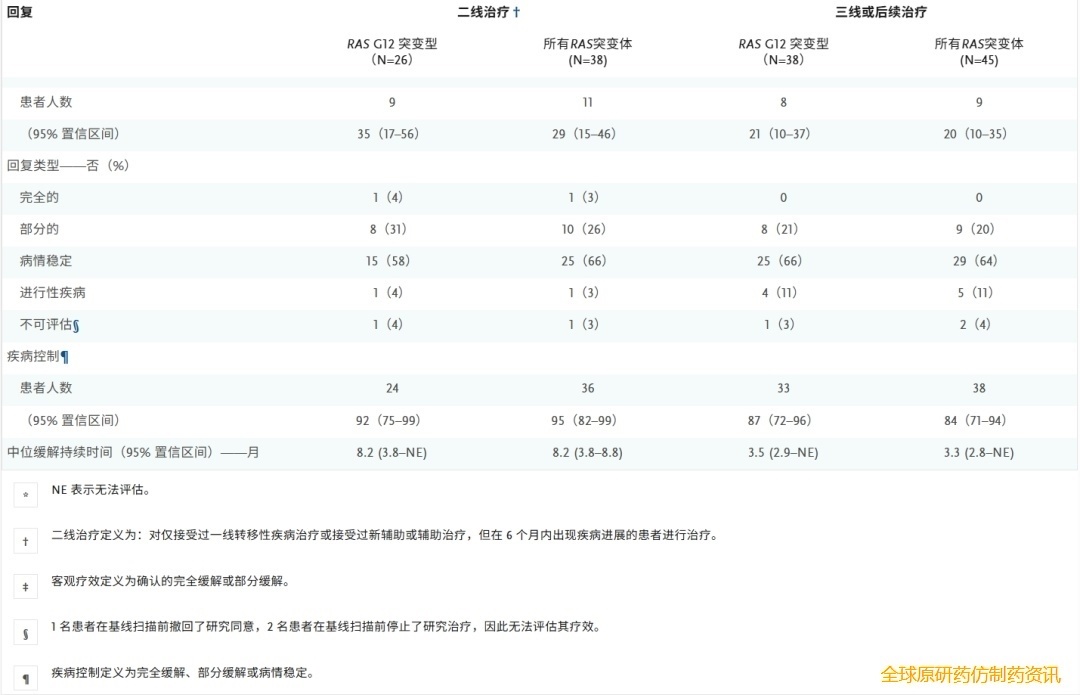
在接受 300 mg daraxonrasib 作为二线治疗的RAS G12 突变患者亚组中,35%(95% 置信区间 [CI],17% 至 56%)的患者对治疗有客观反应。在这些患者中,疾病控制率(计算为完全缓解或部分缓解以及疾病稳定)为 92%(95% CI,75% 至 99%)(图1B和表3)。中位起效时间为 2.6 个月(范围,1.2 至 8.5 个月),中位缓解持续时间为 8.2 个月(95% CI,3.8 个月至无法评估)。
表3. 根据治疗方案,确认最佳总体疗效。
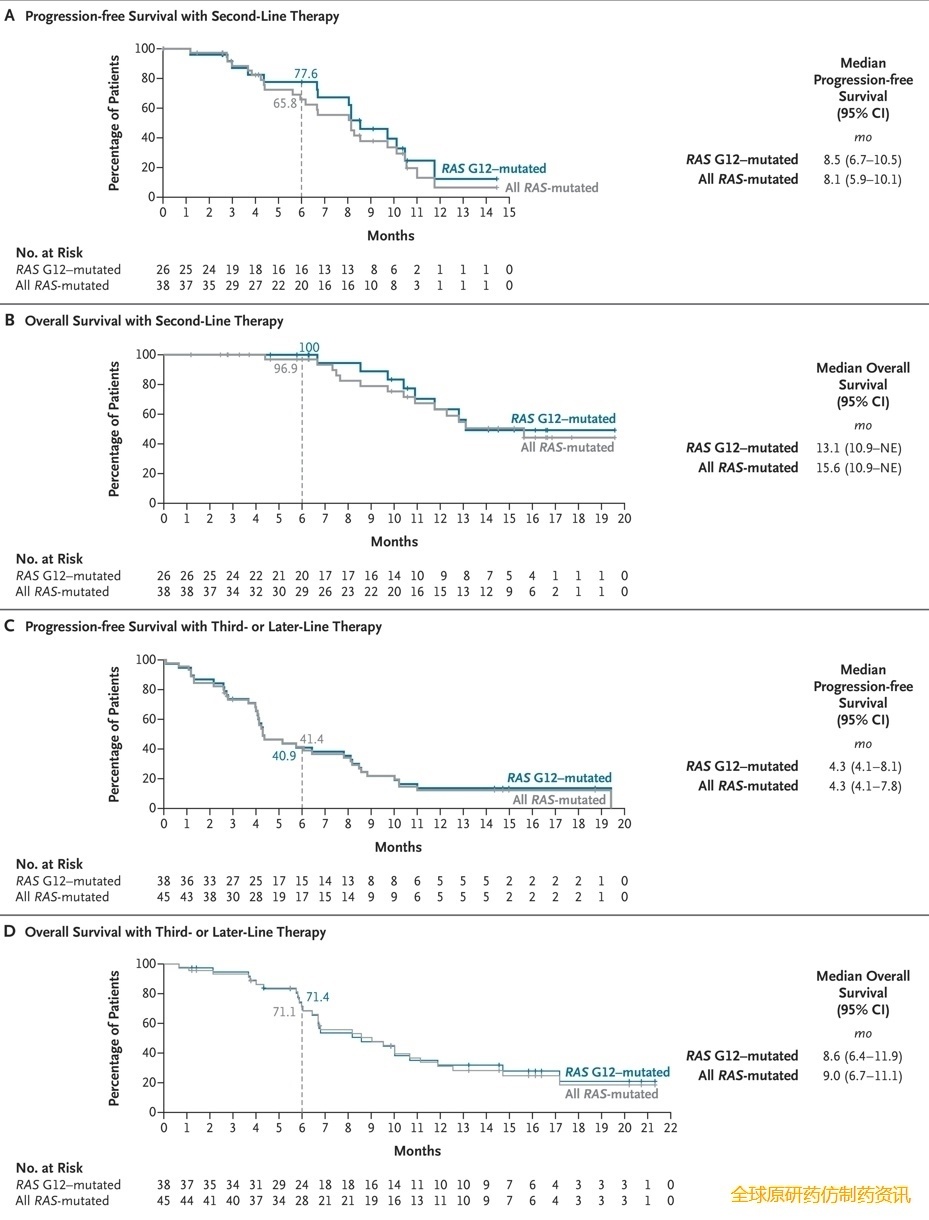
在RAS G12 亚组中,经过 17 个月(范围 10.3 至 24.6 个月)的中位随访后,中位无进展生存期为 8.5 个月(95% CI,6.7 至 10.5 个月)(图2A),中位总生存期为 13.1 个月(95% CI,10.9 个月至无法评估)(图2B)。Kaplan-Meier 估计的无进展生存率在 6 个月时为 78%(95% CI,54% 至 90%),在 9 个月时为 46%(95% CI,24% 至 66%)(表S5);总生存率估计值在 6 个月时为 100%(95% CI,100% 至 100%),在 9 个月时为 89%(95% CI,62% 至 97%)(表S5)。
图2. RAS G12 突变型和所有RAS突变型 PDAC患者的生存情况。
在接受 300 mg daraxonrasib 作为二线治疗的RAS突变患者中,38 例患者中有 11 例(29%;95% CI,15% 至 46%)达到客观缓解,其中包括KRAS G12D、KRAS G12V、KRAS G12R 和KRAS Q61H 突变患者(表3)。达到客观缓解的中位时间为 2.6 个月(范围,1.2 至 8.5 个月)。中位缓解持续时间为 8.2 个月(95% CI,3.8 至 8.8 个月)。中位随访时间为 17 个月(范围 10.3 至 24.6 个月),中位无进展生存期为 8.1 个月(95% CI,5.9 至 10.1 个月)(图2A),中位总生存期为 15.6 个月(95% CI,10.9 个月至无法评估)(图2B)。Kaplan-Meier 法估计的无进展生存率在 6 个月时为 66%(95% CI,47% 至 80%),在 9 个月时为 38%(95% CI,21% 至 55%)(表S5);总生存率估计值在 6 个月时为 97%(95% CI,80% 至 100%),在 9 个月时为 79%(95% CI,59% 至 90%)(表 S5)。图1C、图2C 和图2D以及表 S6显示了接受 daraxonrasib 作为三线或后续治疗的患者的疗效。
目前针对既往接受过治疗的转移性胰腺癌(PDAC)患者的化疗方案疗效有限,缓解率低于10%,中位无进展生存期为2至3个月,中位总生存期为5至7个月。这些方案常伴有3级或4级毒性反应,包括中性粒细胞减少症、腹泻和疲乏——这些副作用通常会导致剂量减少或停药。PDAC中RAS突变的高发生率(>90%)凸显了RAS靶向治疗在拓展PDAC患者治疗选择和改善患者预后方面的巨大潜力。
在1/2期研究中,daraxonrasib 治疗组约三分之一的患者出现3级或以上治疗相关不良事件。大多数治疗相关不良事件为1级或2级,且均为靶向毒性反应,例如皮疹和腹泻,这些不良事件可通过常规临床干预措施得到控制。接受300 mg daraxonrasib 治疗的患者中,近一半因上述不良事件而调整了剂量,但大多数患者能够以相同剂量或降低剂量继续治疗,从而维持了总体剂量强度。没有患者因上述不良事件而停止治疗。建议在未来的试验中采取预防措施,以减少皮疹等3级不良事件的发生。
在 300 mg 剂量下,daraxonrasib 在RAS突变型胰腺导管腺癌 (PDAC)患者中产生了客观缓解和疾病控制。尽管临床前数据表明其对RAS G12 突变具有最强的活性,但目前的临床数据集规模太小,无法确定不同突变体之间的差异。虽然无法进行直接比较,但本研究结果表明,对于携带不同RAS突变的 PDAC 患者,daraxonrasib 的无进展生存期和总生存期估计值均优于既往报道的二线化疗结果。daraxonrasib 通过抑制所有三种RAS亚型、所有关键RAS突变以及处于活性 GTP 结合状态的野生型RAS,与仅靶向非活性 GDP 结合状态的 KRAS G12C 抑制剂的等位基因特异性活性相比,提供了更全面的 RAS 抑制。与已发表的 KRAS G12C 抑制剂在 PDAC 中的研究结果相比,daraxonrasib 似乎显示出更高的缓解率和更长的生存期,但跨试验比较并不可靠。
临床前研究和转化药代动力学或药效学模型证实,胰腺导管腺癌 (PDAC) 的肿瘤消退需要持续的 RAS 通路抑制(≥90%)。在临床前模型中,daraxonrasib 通过优先在肿瘤内蓄积和持久抑制丝裂原活化蛋白激酶 (MAPK) 来达到这一目标。这些发现表明,疗效取决于足够的药物暴露量和通路抑制的维持。转化药代动力学或药效学模型预测,每日一次约 100 mg 的daraxonrasib 可使肿瘤停滞或轻微消退,而每日约 300 mg 最有可能维持通路抑制并最大程度地发挥抗肿瘤活性。群体药代动力学模拟(未发表数据)支持以下预期:每日一次 300 mg 的剂量比在小鼠模型中有效的剂量更有可能达到目标药物暴露量。临床安全性和有效性数据支持每日一次 300 毫克的剂量作为推荐的单药治疗剂量。
daraxonrasib 的抗肿瘤活性反映了其对 GTP 结合型 RAS 的直接抑制作用。RAS 是胰腺导管腺癌 (PDAC) 中的主要信号通路形式,几乎所有 PDAC 肿瘤都携带RAS突变,而最常见的变异体(例如KRAS G12D、G12V 和 G12R)目前尚无获批的靶向治疗。客观缓解率与临床前观察结果一致,即 daraxonrasib 的活性是通过肿瘤内药物积累和暴露依赖性通路抑制,从而强效且持续地抑制 MAPK 通路而实现的。虽然 daraxonrasib 的耐药机制仍在研究中,但其疗效的持久性可能反映了其对包括野生型 RAS 在内的多种 RAS 变异体的选择性活性,这可能减轻了由继发性突变或其他致癌改变引起的代偿性信号传导所导致的耐药性,而这些耐药性已在等位基因选择性抑制剂中有所报道。治疗指数似乎得到了以下因素的支持:对活性 GTP 结合 RAS 的选择性、PDAC 对持续通路信号的依赖性、以及相对于正常组织的优先肿瘤内积累——所有这些都使得临床上有效的暴露成为可能。
由于这是一项 I/II 期研究,因此应谨慎解读结果。较小的亚组样本量限制了抗肿瘤活性评估的精确性,以及基于等位基因或共突变状态的解读。单组设计也使得无法与标准治疗方案进行明确的比较。尽管如此,临床活性、快速且持久的疗效以及主要为低级别不良事件的持续证据支持在针对晚期胰腺癌患者的随机试验中进一步评估 daraxonrasib。这些结果支持正在进行的 RASolute 302 研究,这是一项全球随机 III 期试验,旨在比较 daraxonrasib 与化疗作为转移性胰腺癌患者二线治疗的疗效(ClinicalTrials.gov 编号:NCT06625320)。
在接受过RAS突变 PDAC治疗的患者中,daraxonrasib 显示出抗肿瘤活性,但三分之一的患者出现了 3 级或更高级别的治疗相关不良事件。