肿瘤疫苗是通过将特异性抗原呈递至抗原提呈细胞,随后刺激免疫系统激活并诱导机体产生细胞免疫和体液免疫应答以获得抗肿瘤免疫能力,进而控制肿瘤细胞增殖、转移和侵袭等。根据递送的种类主要分为蛋白质类疫苗、细胞疫苗和核酸类疫苗。2025年2月5日Nature杂志发布了一项个体化癌症疫苗(PCV)的最新研究结果。
该研究由哈佛医学院附属医院丹娜-法伯癌症研究所牵头的I期临床研究。纳入了9名高风险(III/IV期)肾细胞癌患者。患者在肿瘤完全切除后,接受了靶向新抗原的PCV治疗。这些PCV由包含多种新抗原的长多肽链组成。其中5名患者同时接受了Ipilimumab的辅助治疗,另外4名患者只接受了PCV治疗。长期随访结果显示,在中位随访时间为40.2个月时,所有患者均未出现癌症复发。其中两名患者的随访时间已经达到4年。安全性方面,PCV的安全性和耐受性良好,最常见的不良事件为低级别的注射部位反应和暂时的流感样症状,没有患者出现3级以上毒性。
美国约翰斯·霍普金斯医学院Yarchoan在2024年AACR年会上报告并同时发表在《自然医学》上的研究显示,为肝癌患者量身定制、编码至多达40个肿瘤新抗原的个性化癌症治疗疫苗GNOS-PV02,与PD-1抑制剂联合用于肝癌患者的后线治疗时,仍有着良好的抗肿瘤活性和出色的安全性,其中3例患者达到了CR!该研究纳入了36例已接受过仑伐替尼/索拉非尼治疗的晚期肝细胞癌(HCC)患者,主要终点是安全性和免疫原性;次要终点为客观缓解率(ORR)、无进展生存期(PFS)和总生存期(OS)。探索性终点包括评估肿瘤和免疫生物标志物及其与治疗结果的关联。结果显示:在进行至少一次治疗后重新评估的34名患者中,根据RECIST 1.1评价标准,客观缓解率为30.6%(11/36),完全缓解率为8.3%(3/36),部分缓解率为22.2%(8/36),疾病控制率为55.6%(20/36)。中位随访时间为21.5个月,对于有反应的患者,中位反应时间为9.3周(范围8-46周)。中位缓解持续时间未达到。中位无进展生存期为4.2个月,中位总生存期为19.9个月。值得一提的是,目前中位总生存期数据优于既往PD-1抑制剂单药二线治疗肝癌的数据(12.9-15.1个月)。一名患者在接受五次PTCV后,原本不可手术的HCC达到了二次可手术性,并在首次治疗剂量后保持了18.2个月的无肿瘤状态。GNOS-PV02是一种个性化治疗性癌症疫苗(PTCV)。其作为一种创新的癌症免疫治疗策略,旨在针对每位患者肿瘤中特有的突变相关新抗原(MANAs)来设计和制造。这种方法的核心在于利用患者自身肿瘤的基因信息来定制疫苗,使之能够激发患者免疫系统的特异性反应,针对那些仅在肿瘤细胞上表达的新抗原,从而识别并杀死肿瘤细胞,同时最大限度地减少对正常细胞的影响。新型个体化癌症疫苗LK101注射液是一种国产化个性化的新抗原靶向癌症疫苗,也是一种基于树突状细胞 (DC) 的 mRNA 疫苗。2024年ASCO年会上公布了其首次人体试验的惊艳结果。
研究共有24名HKLC IIa期HCC患者按1:1的比例分配到消融对照组和消融联合DC疫苗(LK101)组。结果显示:疫苗接种组和对照组患者的中位随访时间为48.4个月和38.8个月。消融对照组有3名患者死亡,而所有接种疫苗的患者均存活。这意味着,12名接受疫苗的患者生存期长达4年以上。
两组患者的1年和2年复发率分别为18.2%vs.33.3%和36.4%vs.51.4%。总体而言,所有患者均引发了至少一种可测量的反应。患者接种疫苗后,对60-90%的肿瘤新抗原肽产生免疫反应。此外,接种疫苗后患者外周血中CD4和CD8阳性效应记忆T细胞均呈增加趋势,PD-1阳性T细胞减少。
2024年美国癌症研究协会(AACR)年会上,公布了个体化癌症疫苗Autogene cevumeran (BNT122)治疗胰腺导管腺癌(PDAC)的临床试验随访成果。该项研究是由研究者发起的单中心Ⅰ期临床试验,旨在评估辅助Autogene cevumeran联合阿替利珠单抗和改良FOLFIRINOX(亚叶酸钙、氟尿嘧啶、伊立替康和奥沙利铂)用于可切除胰腺癌患者的安全性和免疫原性。2023年5月,国际顶尖学术期刊 Nature发表了该试验的研究结果。
该研究共纳入34例PDAC患者,其中28例接受了手术治疗。术后,19例患者接受了阿替利珠单抗(Atezolizumab)治疗,其中16例随后接种了BNT122疫苗,而其中15例患者还接受了mFOLFIRINOX治疗。在16例可评估的患者中,8例患者的治疗是可耐受的,并且诱导了免疫应答(通过高强度新抗原特异性T细胞扩增来测量)。此外,在中位1.5年的随访中,8名无应答者的中位无复发生存期(RFS)为13.4个月。此外,一名患者的血清CA19-9水平升高,并出现了新的微小肝脏病变(约7mm),但在随后的成像中,该病变消失。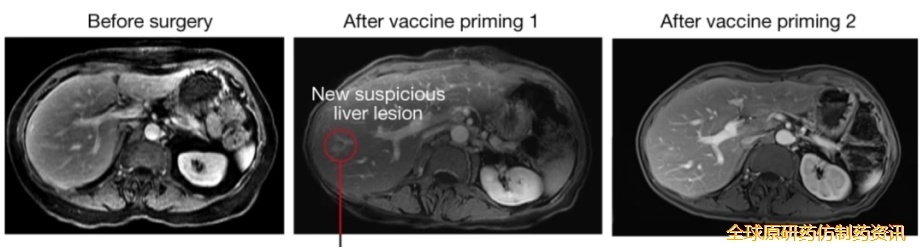
接种BNT122疫苗前后患者腹部MRI对比
2024年AACR年会上,研究者报告了最初研究的长期随访结果。经过中位3年的随访,8例疫苗诱导T细胞应答患者的中位无复发生存期(未达到)明显长于未出现免疫应答的患者(13.4个月)。并且,其中的6例在接种疫苗3年后分离的CD8+ T细胞对靶向新抗原仍有反应,保持无病状态。
一项旨在比较Autogene cevumeran联合阿替利珠单抗和FOLFIRINOX对比标准治疗FOLFIRINOX在可切除胰腺癌患者中的疗效和安全性的随机Ⅱ期临床试验,正在进行中。计划在全球近80个地点招募约260名患者。
(4)前列腺癌
一项Ⅰ期临床试验中共12例去势抵抗性前列腺癌(CRPC)患者接受了256μg( n=3)、640μg(n=3)或1280μg(n=6)的CV9103(mRNA疫苗)治疗,在第1、3、7、15和23周分别经皮内给药。Ⅰ期临床试验确定的推荐Ⅱ期剂量为1 280μg。Ⅱ期临床试验中,共有44例CRPC患者接受了1280μg CV9103治疗。主 要终点为安全性,次要终点是免疫原性。
44例CRPC患者的中位总生存期(mOS)为 29.3 个月,转移性CRPC患者亚组( n=36)的mOS为31.4个月。在26例可通过体外酶联免疫吸附法等技术评估免疫反应的转移性CRPC患者中,对CV9103有反应的患者(n=20)与无反应者(n=6)相比,具有更好的临床获益, 对 CV9103 产生的反应性抗原数量多的亚组较数量低的亚组生存更具有优势。
(5)恶性黑色素瘤
BNT111正是一种即用型治疗性疫苗,它能编码4种非突变的肿瘤相关抗原(NY-ESO-1、MAGE-A3、酪氨酸酶和TPTE)。这些抗原在超过90%的皮肤黑色素瘤中表达。美国FDA在2021年授予BNT111快速通道资格。
BTN111在其前期的Ⅰ期试验(LipoMERIT研究)中,显示了良好的效果。该研究共入组了115 例至少表达上述1种抗原的ⅢB/C及Ⅳ期黑色素瘤患者。采用初始/重复强化方案静脉注射BNT111。
患者被分为7个剂量递增队列(目标剂量范围为7.2~400.0μg)和3个剂量扩展队列(14.4μg、50.0μg和100.0μg),旨在研究BTN111的疗效和安全性。
入组患者被分组入有疾病证据(ED)组或无疾病证据(NED)组来评估有效性和安全性,并通过干扰素 -γ 酶联免疫吸附法分析免疫原性。
结果显示:所有剂量组均出现肿瘤的消退,大多出现部分缓解(PR)或者疾病稳定(SD)的患者都能展现出持续的疾病控制,部分患者疾病控制时间长达 2 年多。NED 组患者临床疗效良好,中位无病生存期(mDFS)为34.8个月。
安全性方面两组患者相似,主要为轻至中度发热或寒战等类似流感的表现,大多早发、短暂且可使用退热药控制。
2024年7月31日,公布BNT111的2期临床试验积极顶线数据。分析显示,试验达成主要终点,BNT111联合PD-1抑制剂Libtayo(cemiplimab)联合疗法能够显著改善无法切除的晚期黑色素瘤患者的总缓解率(ORR)。

2023年12月14日,Moderna和默沙东公布了mRNA疫苗与帕博利珠单抗临床Ⅱ期试验的最新数据,其惊艳的研究数据使之有望成为首款上市的mRNA肿瘤疫苗,最早或将于2025年问世!KEYNOTE-942研究数据显示,联合研发的基于患者肿瘤DNA的个体化治疗mRNA-4157/V940疫苗,显著改善了患者的无复发生存期(RFS),将高危Ⅲ/IV期黑色素瘤患者的复发或死亡风险降低49%,远处转移或死亡风险降低62% 。不良反应方面,联合治疗组较单药治疗组并未发现新的不良事件。mRNA癌症疫苗mRNA-4157/V940联合Keytruda组合疗法获美国食品药品监督管理局(FDA)授予突破性疗法认定,用于高危黑色素瘤患者接受完全切除术后的辅助治疗。(6)非小细胞肺癌
CIMAvax-EGF世界上首款针对非小细胞肺癌的治疗性疫苗。2011年完成了临床试验正式在古巴上市,数据显示:该疫苗的使用延长了患者的寿命,从6个月到5年不等。2023年的ASCO大会上,美国公布了CIMAvax-EGF和Nivolumab 作为铂类化疗后二线 (2L)治疗晚期非小细胞肺癌 (NSCLC) 的Ⅱ期试验的最终结果。研究共入组了23名晚期NSCLC患者,在铂类化疗后作为维持疗法,患者每两周接受一次CIMAvax-EGF和Nivolumab治疗。结果显示:在21名可评估的患者中,疾病控制率(DCR)为 47.6% (n=10);3 年总生存(OS)率为 29%;中位总生存期OS为11.9个月。其中,鳞癌患者具有更高的3年总生存率为达到50%。这意味着,一半的晚期肺鳞癌患者生存时间达到了3年。(7)胶质母细胞瘤
2024年5月1日,Cell杂志发表一项题为“RNA aggregates harness the danger response for potent cancer immunotherapy”的研究中,来自佛罗里达大学的科学家们报道了以新型mRNA疫苗治疗胶质母细胞瘤的积极进展。在针对4例成人患者的首次人类临床试验中,研究团队开发的mRNA疫苗通过静脉注射迅速激发了免疫系统对肿瘤的强烈排斥反应。
研究者们利用患者自身的肿瘤细胞来制造个性化mRNA疫苗,且使用了一种新的复杂递送机制。具体来说,科学家们开发了“洋葱样”多层RNA脂质颗粒聚集体(lipid particle aggregates,LPAs),以大大增强肿瘤mRNA抗原的载荷包装和免疫原性。
结果显示:在首批接受治疗的4名成年患者中,治疗效果良好,包括重新编程TME、引发胶质母细胞瘤患者的适应性免疫等。最令人印象深刻的是,该疫苗在注射后48小时内,即可将“冷肿瘤”迅速转变为“热肿瘤”,从而产生快速且持久的抗癌免疫反应!目前,这些患者的生存期和无病生存期都已超过了预期!
(8)其他实体肿瘤(头颈部肿瘤、结直肠癌等)
Keynote603研究旨在评估mRNA-4157单独治疗已切除原发肿瘤的实体瘤患者(A 部分)和联合帕博利珠单抗治疗晚期/转移性实体瘤患者(B部分)的安全性和有效性。
Ⅰ期临床试验中,13例已切除原发肿瘤的实体瘤患者均完成 mRNA-4157 肌内注射最多9个周期的治疗,其中11例患者在接受第1剂mRNA-4157后75周(近19个月)内没有出现疾病复发,且未观察到 3 级及以上TRAEs发生。
扩展队列的中期分析结果显示,63例联合治疗患者中:
① 3例患者(1例头颈部鳞状细胞癌, 1 例结直肠癌和1例前列腺癌)达到了完全缓解(CR);
② 8例患者(4例头颈部 鳞状细胞癌、2例小细胞肺癌、1例膀胱癌和 1例子宫内膜)达到了PR。
③10例人乳头瘤病毒(HPV)阴性头颈部鳞状细胞癌患者中,客观缓解率(ORR为 50%,(1例 CR,4例PR,4例 SD),mPFS为9.8个月,这与帕博利珠单抗单药的效果相比是有利的。
安全性方面:所有剂量 mRNA-4157 均可耐受,未出现 DLT,未有3/4 级TRAEs及SAEs发生。
浙江邵逸夫医院研究团队设计了一种基于肽的新生抗原疫苗iNeo-Vac-P01,并进行了一项前瞻性临床试验,共纳入22例标准治疗失败的泛癌种晚期实体瘤患者。利用iNeo人工智能疫苗设计平台、个体化多肽药物制备及质控体系为每例患者定制了iNeo-Vac-P01疫苗。结果显示:22例晚期恶性肿瘤患者中,20例无不良反应发生或有轻微不良反应。个体化肿瘤疫苗对于泛瘤种实体肿瘤,疾病控制率达到71.4%,中位无进展生存期为4.6个月而中位生存期中位数未达到。研究表明,对于晚期实体瘤患者来说,iNeo-Vac-P01单药治疗是可行且安全的。它可以诱导T细胞介导的针对肿瘤新生抗原的免疫应答,可能具有良好的抗肿瘤疗效。相比于其他免疫治疗方案,mRNA疫苗具有以下优势:
1.治疗机制独特:除了编码抗原的mRNA能够刺激针对肿瘤的特异免疫应答外,mRNA本身也具有固有的免疫刺激特性。通过合理调节mRNA的免疫原性,可以使其充当佐剂,提高疫苗的免疫效果。2、开发周期短:相比于CAR-T、TIL等细胞疗法,mRNA疫苗的开发周期较短。3、安全性高:相较于DNA疫苗和病毒载体疫苗,mRNA疫苗不会进入细胞核,因此不会导致潜在的基因组插入突变风险;同时mRNA在体内可以通过自然途径进行降解,不会在体内积累。










